仿制药制剂出口在当下已成为国内药企的重要意识,越来越多的制药企业开始将业务拓展到海外。一方面海外业务为国内药企带来可观收入,另一方面被美国或欧盟批准的制剂产品使药企拿到绿色通行证,获得更快的国内报批资格。那么制剂出口需要建立怎样的体系?研发及生产流程是怎样的模式?
以美国市场为例,仿制药制剂产品需要在FDA(美国药监局)认证的cGMP(动态药品生产管理规范)条件下生产,并具有与原研产品较高的体内外一致性,才有资格获批进入美国市场销售。也就是说,首先,药企需要拥有一个FDA认证过的cGMP车间;其次是关注产品质量,开展体内外一致性评价研究;最终目标是获得与原研产品体内生物利用度一致的仿制药产品,也就是我们常说的通过BE(生物等效性)实验。
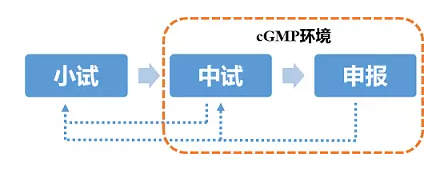
产品的研发从实验室小试,到车间中试放大,再到申报生产,是一个较为漫长且需经过不断反复循环的过程,通常经历3-5年时间。
实验室小试阶段
批量通常在200g~2kg
当仿制药产品立项后,需要对原研产品进行较为全面的调研及检测工作。其目的在于明确研发目标,使研发的每一个环节的目标具体化、量化及可控化。
调研工作包括:原研产品的新药保护期信息、FDA橙皮书中收录的关于该产品的专利保护内容、原研产品的制剂组成、相关专利提供的可能的制剂制备方法、以及FDA推荐的溶出方法和BE实验方案等;
检测工作包括:原研产品的外观尺寸、药物含量、杂质及药物溶出曲线等测定数据。
对原研产品的处方及工艺越了解,则越有利于仿制药的研发。然而,FDA明确披露的只有处方中的辅料类别,即产品由原料药与哪些辅料组合而成。而对于每种辅料所占的确切比例,以及制剂的实际制备工艺并无介绍,通常需要通过一些相关专利或文献的信息披露推测获得,这也是仿制药产品要摸索的最关键的环节之一。如果专利中明确保护了某种辅料或某个工艺,那么在专利过期前需要规避并选择其他解决方案,这就使仿制药的制剂研发难度大大提高。
对原研产品理化性质的测定帮助我们更好的理解目标性状。对于口服固体制剂来说,通常在溶出实验中具备与原研片一致性较高的产品才有更高的BE实验通过率。因此在这一阶段,我们最关注的是所研发仿制药的溶出曲线与原研产品的匹配情况,而如何开发体内外相关性高的溶出方法,更是BE实验获胜的关键。
在实验室小试阶段,通过对处方及工艺的筛选及优化,获得与原研产品溶出度非常接近的产品,并明确处方中的关键辅料及工艺中的关键参数对溶出曲线的影响趋势。值得注意的是,搞清楚影响趋势,要比“碰运气”找到了最佳溶出曲线重要得多。
中试放大阶段
批量通常在20kg~50kg
优化获得了与原研产品较为接近的备选处方后,即可进入车间中试放大阶段。通常从这一阶段开始到未来申报生产及商业化生产等均要求在cGMP环境下操作。
从小试到中试,是批量变化最大的一步,也是最容易产生“放大效应”的一步,处理好放大效应是产品最终走向申报乃至商业化生产的成功关键。
放大效应有两个方面:一是参数的设计问题,即如何参照小试的设备工艺参数来设定中试放大中大设备的工艺参数。打个比方说,煮1个鸡蛋如果需要10分钟,那么煮20个鸡蛋通常不只是相同的10分钟,也一定不需要等比扩大20倍后的200分钟。因此,推算并确定中试放大的关键工艺参数是一个难度较大的问题。
另一个容易出现的放大效应是,有些问题在小试实验中不存在或很小,到中试生产才显现出来并存在严重缺陷,也就是放大生产可行性的问题。例如,一些产品在实验室小试中并无压片问题,但在中试生产高速旋转压片时可能出现物料粘冲、分层、可压性差的问题,这些问题只有在批量较大时才会显现,因此即使小试研发的产品质量再好,在未来也是无法量化生产的。
在解决了上述“放大效应”问题后,我们来关注产品质量。大部分产品在研发时,相同的处方及工艺在小试和中试放大分别得到的产品的溶出曲线存在一定差异,通常会返回实验室进行调查研究,以及重复放大生产进一步优化参数设计。当然,如果在小试研发阶段对溶出曲线变化趋势的影响因素研究的十分清晰透彻,也可更快更准确地使产品接近目标值。
这样的交叉循环反复工作可能持续半年到两年的时间,甚至更久,视产品研发难度及放大效应大小而定,最终目标是获得与原研产品体外各方面指标一致且质量可控的中试产品。
申报生产阶段
批量通常在50kg~150kg
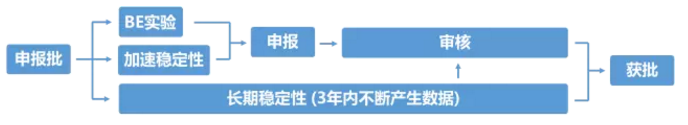
申报生产是整个研发过程中最为关键的一个环节,也是申报材料中FDA最关注的生产批次。目前FDA的要求是,须连续生产三批且每批批量不小于10万片(粒)的产品。这里的连续,并非中间一天不能停歇,而是中间不能再穿插任何放大生产批次。
由于申报批的批量与中试批量相差不多,为了节省时间及成本可视研发难度等情况考虑是否在申报批前做1-2批与申报批次同等批量的放大研究。
三批申报产品生产完成后,需要同时开展长达6个月的加速稳定性实验和长达3年的长期稳定性实验,只选择其中一批进行BE实验(通常是第一批),并撰写及整理申报材料。6个月加速稳定性实验结束后,如果检测结果符合标准,且已通过BE实验,则这三批申报产品具备申报资格,可以向FDA递送材料进行申报。材料递送后,FDA还需要1-2年审批时间,中间会提出一些缺陷性问题,药企需回答问题,并可能还需要补充小试或中试实验数据,同时补充不断获得的长期稳定性实验数据。
在申报生产环节,最为关键同时也是难度最大的一个步骤是BE实验。只有BE实验成功的申报批产品,才意味着在体内的释放及吸收行为与原研产品完全一致。美国市场仿制药BE实验的成功率仅为48%左右。一旦BE失败,三批申报产品全部报废,研究者要对失败原因进行全面的调查和评估。
通常情况下,如果BE实验结果与原研产品相差甚微时,有以下两种途径快速重新完成申报工作。①如果仅仅是在生产过程中工艺参数控制不当,可在设置范围内做细微调整,并立即重新开始三批申报工作;②如果需要调整的工艺参数超出生产文件设置范围,则需要修改升级生产文件后,再进行申报生产。
如果BE实验结果与原研产品相差较大时,很可能就要返回到最初的实验室小试阶段重新开始研究。
可能有人会问,体外溶出实验中申报产品已与原研产品具有较高的溶出匹配度,为什么体内BE实验还会失败?一般来说,未找到更具区分性或与体内相关性更好的溶出方法是BE失败的主要原因。因此实验室小试研究需要这可能是因为缺乏深入全面的体外溶出实验研究。着重确定最佳的溶出分析方法,并在新方法下重新优化处方及工艺。
结语
制剂出口海外市场前景广阔,但对制剂质量的要求也极高,所需研发周期较长,成本较高,因此对于药企来说,需要具备丰富的制剂技术经验与资本积累,才有可能生存下去。这也极大推动了中国药企制剂水平的飞速发展,使市场建立起优胜劣汰的健康的竞争模式。



























 相关新闻
相关新闻

 关于我们
关于我们