豁免来由
体内生物等效性BE的研究是证明制剂之间治疗等效性的一种手段,主要通过测量血药浓度来判断药效是否等效。BCS的概念在提出之前,申请者一般是很难有一个科学的理论作为依据向相关的监管部门提出豁免BE;今年来,随着BCS概念的不断提出和不断论证,越来越多的监管机构开始考虑并接受基于BCS的生物等效性豁免,如FDA、EMEA、WHO。
BCS全称为biopharmaceutics classification systm,国内一般译为“生物药剂学分类系统”,是根据药物在水中的溶解度和肠壁渗透能力对药物进行科学分类的标准。对于口服固体速释制剂而言,原料药的溶解性、渗透性和制剂的溶出度对这3个方面基本决定了药物在体内的吸收速度和程度:BCS I:高溶解性 & 高渗透性;BCS II:低溶解性 & 高渗透性;BCS III:高溶解性 & 低渗透性;BCS IV:低溶解性 & 低渗透性。
如果体外研究能充分证明体内性能无差异,那么可豁免体内生物等效性的研究。对于BCS I类药物而言,如忽略制剂中辅料的影响,通常可免除体内生物等效性研究;BCSIII类药物在体内吸收的限速步骤是药物的吸收,如药物在体内能迅速释放,并能在到达药物吸收部位时达到饱和浓度,则可以忽略不同制剂释放速率的差异。因此,目前寄希望更多于BCS I和BCS III药物的豁免。
西咪替丁基本介绍
西咪替丁属于组胺H2受体拮抗剂,长期用于治疗十二指肠溃疡、胃溃疡及卓-艾综合征。西咪替丁于1971年被发现,1977年开始被商业化使用,原研公司为GSK(但2002年均撤市),已有较长的使用历史。
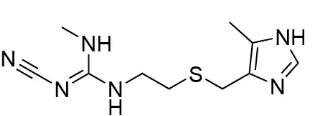
西咪替丁化学结构式
BCS分类:在pH1-8的介质中(37度),西咪替丁的溶解度均能达到6mg/ml;市售最大规格为0.8g (CFDA批准的最大规格为0.2g),即约用133ml水即能将其溶解(小于250ml),所以西咪替丁归属为具有高溶解性;通过对健康受试者测试,西咪替丁在体内的绝对生物利用度约为60%(低于85%),故将其归属为低渗透性。根据BCS分类概念,西咪替丁属于BCS III类药物。
安全性:西咪替丁属于治疗窗宽的药物,健康成年人肾功能正常时半衰期(t1/2)约为2小时。关于西咪替丁的副作用比较少,被报道的有腹泻、皮疹、头昏眼花、疲劳、便秘、肌肉痛等,这些现象通常都比较轻微、发作时间也比较短。总的来说,几乎没有什么副作用引起重大临床事件。西咪替丁的血药浓度可通过反相HPLC有效检测。
自研与原研在体内外的差异
西咪替丁片的原研公司为GSK,1977年获得FDA批准的NDA,商品名为TAGAMET®,批准规格为200mg、300mg、400mg、800mg;同时还获得盐酸西咪替丁注射剂的NDA,活性成分为盐酸西咪替丁,批准规格为300mg/2ml。
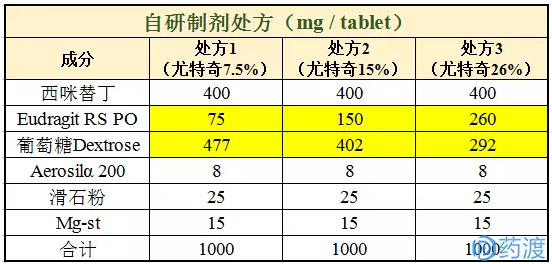
方案设计:在自研处方中,在保持片重不变(1000mg)的情况下,通过调整处方中尤特奇Eudragit(缓释材料)与葡萄糖的比例来刻意造成体外溶出曲线的巨大差异,最终与原研制剂产品进行体内PK比较。
自研处方与原研的体外行为
以西咪替丁片400mg为例,每片中含有西咪替丁400mg。体外溶出曲线的差异主要通过尤特奇Eudragit RS PO控制;三个体外溶出有明显差异的成品最终在健康受试者体内进行相对生物利用度的研究。
自研产品均采用直压工艺,所有的原辅料经过筛后混合后采用单冲压片机压制。各项检测指标如片重差异、硬度、含量均匀度均符合现行的药典要求。
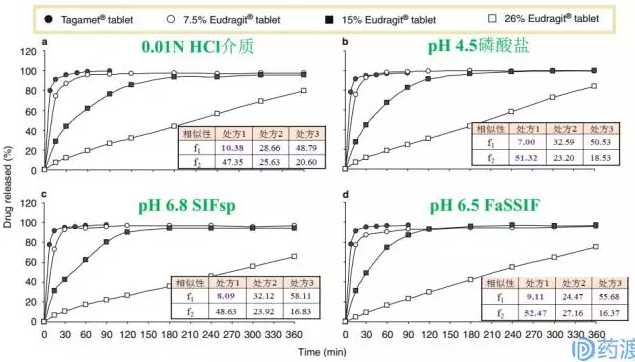
溶出条件:①0.01N HCl介质,pH约为2.0;②pH 4.5的磷酸盐缓冲液;③不含酶的人工肠液(药典配制方法),pH约为6.8;④FaSSIF(空腹状态下模拟肠液),pH约为6.5,其中,第一、二、三种介质的选择是根据BCS豁免指南指定介质来的,第四种介质FaSSIF模拟液是基于胆盐的考虑而额外增加的。溶出介质500ml,桨法75rpm。将自研的三个制剂与原研制剂Tagamet-400mg分别在这四种介质进行溶出速率测试,分别测定它们在不同pH条件下的溶出曲线;同时,通过溶出曲线突出自研三个制剂的体外差异。最终均将其与原研数据比较并计算差异因子f1和相似因子f2。
取样点为15min、30min、60min、90min、120min、180min、240min、300min、360min
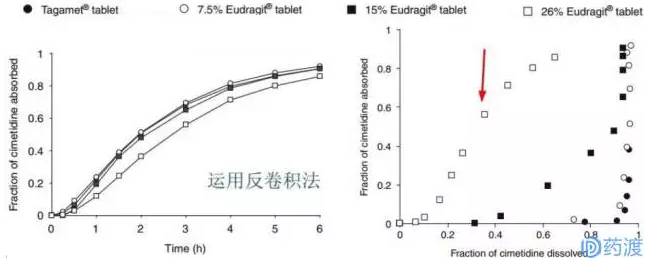
从上述曲线中可看出:
原研制剂在上述四种介质中15min溶出均低于85%,30min则高于85%;
三个处方和原研制剂分别在四种不同pH介质中呈现的溶出曲线是相似的,说明在它们在胃肠道的上段的环境中具有相似的溶出趋势;
7.5% Eudragit(处方1)在四种介质中30min内溶出达到85%,差异因子f1均符合相似性要求(<15),但相似因子f2有的介质符合要求,有的介质不符合要求(≥50),因此,从体外溶出曲线上看,处方1与原研处于相似的边缘;
15% Eudragit(处方2)在四种介质中在120min处溶出才接近85%,而26% Eudragit(处方3)在360min处溶出才接近80%;
处方2和处方3与原研制剂对比的f1和f2均不满足相似性的要求,且均是在30min后溶出才达到85%。
因此,从体外溶出上看,似乎处方1勉强能与原研相似,处方2和处方3则不能称与原研相似。
自研处方与原研的体内行为
方案设计:征集12个健康的志愿者(其中2个为候补)采用单剂量给药、5种方式的Williams设计方法。志愿者样本数量是根据文献报道数据然后在PS网站(PS:Power and Sample Size )中计算而来的,平均年龄21.7岁 (20 - 24岁),平均体重59.5kg (52.5 - 67.5kg)。所有志愿者在空腹过夜后(不超过10小时)首次给药,或者服用400mg的片剂、或者被注射300mg/2ml的原研注射剂。服用片剂的志愿者在服药后服用一玻璃杯水(约250ml),四小时后服用标准餐。服用片剂的志愿者,在0min(服药前)、30min、60min、75min、90min、105min、120min、150min、180min、210min、240min、360min、480min、600min和720min处分别收集血液5ml;而对于处方2和处方3的制剂,还需在960min和1444min处收集血液。注射剂志愿者注射药物后在0min(服药前)、10min、20min、30min、45min、60min、75min、90min、120min、150min、180min、210min、240min、360min和480min处分别收集血液。试验符合《赫尔辛基宣言》要求。
借助软件,通过血药浓度评估以下PK参数:
AUC12:0到12小时血药浓度-时间曲线下的面积;
AUCo (ug h/ml):0到无穷大时间血药浓度-时间曲线下的面积;
F (%):绝对生物利用度;
Cmax (ug/ml):血药最大浓度;
MRT (h):mean residence time,平均滞留时间。
试验结果
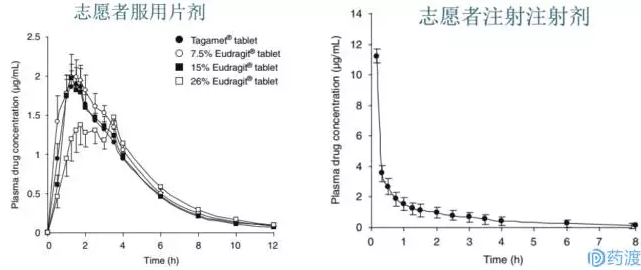
下图为分别为通过服用片剂(左图)和注射注射剂志愿者(右图)的血药浓度-时间的曲线图:
注:每个点代表12个志愿者血药浓度的平均值
从上图左图中可看出,所有片剂处方在口服12小时的吸收情况即可反应药物在体内的整个吸收情况,虽然处方2和处方3还有取样点960min (16h)和1440min (24h),但12小时后的血药浓度量均太低,低于色谱条件的检测下限。因此可忽略。从左图还可发现,处方3 (26% Eudragit)有三峰现象,而已有报道称西咪替丁吸收会有双峰或多峰;但处方3的Cmax显著慢于其它处方,吸收曲线与其它处方有较大差异。
通过IVIVC分析发现,除了处方3 (26% Eudragit)外,其它处方均表现为药物渗透性是体内吸的限制因素;而处方3呈现的是线性关系(R2=0.9685),见下图:
因此,本案例可间接说明,只有当西咪替丁体外溶出120min内未达到85%,才会影响到体内的吸收速率,即西咪替丁120min内体外溶出达到85%则不会影响到体内吸收。
计算机模拟分析
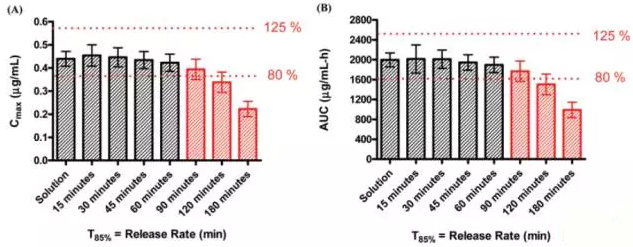
Yasuhiro Tsume and Gordon L. Amidon在2010年借助Gastroplus模拟分析了部分药物的体外溶出与体内吸收的情况,下图为西咪替丁的模拟结果:
作者主要考察溶出达到85%时所需的溶出时间。上图的每个数据是基于24个虚拟试验的平均值而获得的(即假设受试者24例),其中,黑色框框表示预测具有生物等效性,红色则表示体内不等效。通过模拟可发现,药物体外溶出60min内达到85%,体内基本上是等效的;而在空腹时,胃排空时间一般为15-60min。
下图为模拟西咪替丁体外溶出在15min达到85%时体内药物吸收情况,其中,红色线条代表体外药物释放量-时间的曲线;蓝色线条代表体内药物吸收量-时间的曲线。
上图中,可明显看出当西咪替丁体外溶出度在不到1小时即达到平台期,其体内吸收仍需3~4小时才完全吸收完,因此,限制西咪替丁体内吸收的关键因素为其较差的渗透性,而非体外的溶出速度。
小结
假如药物中的辅料既不会影响胃肠道上段的蠕动,也不会影响活性成分的体内吸收,且药物在所有生理pH条件下均具能快速溶出,那么,这些BCS III药物在体内具有相似的行为。事实上,目前已有多个BCS III类药物被报道其体外溶出不一致但体内一致,因此它们被推荐为豁免药物的候选者,但似乎很少有研究能直接证明当药物溶出多慢时才会影响到这些III类药物的吸收,而非仅仅局限于“15min内达到85%”这个“极快速溶出”条件。
本研究所用的辅料均为FDA批准的辅料及对应的用量(IID中可查询),通过外在条件制造西咪替丁体外极快速溶出(处方1)和体外缓慢溶出(处方3)证明了西咪替丁在体外溶出120min未达到85%(处方2的限)才会影响体内吸收。因此,若不考虑辅料的影响,西咪替丁速释制剂获得豁免的可能性非常大;我们只需要的辅料的选择上多加小心。
国内情况
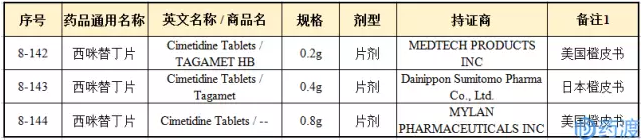
2017年07月21日,CFDA发布的仿制药参比制剂目录(第八批)通告指出,西咪替丁片的参比制剂如下:
CFDA网站显示,共有388个西咪替丁单方片剂产品和213个西咪替丁单方胶囊产品获得CFDA的批准。根据药智数据统计,目前有10条参比制剂备案信息,涉及企业7家,均为单方片剂,具体见下表:
主要参考资料:
1.Feasibility of Biowaiver Extension to Biopharmaceutics Classification System Class III Drug Products Cimetidine
2.The Biowaiver Extension for BCS Class III Drugs: The Effect of Dissolution Rate on the Bioequivalence of BCS ClassIII Immediate-Release Drugs Predicted by Computer Simulation
3.Clinical pharmacokinetics of cimetidine
4.FDA、WHO和EMA关于基于生物药剂学分类系统的生物等效性豁免指导原则的比较
5.人体生物等效性试验豁免指导原则





























 相关新闻
相关新闻

 关于我们
关于我们